
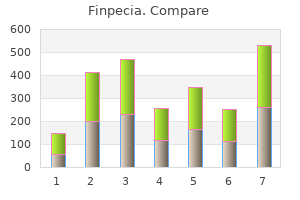
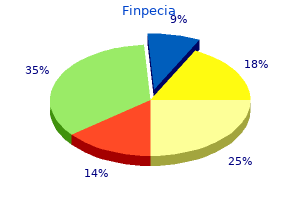
Finpecia
"Buy online finpecia, hair loss in toddlers".
By: Q. Taklar, M.A., M.D., M.P.H.
Deputy Director, Harvard Medical School
Nonetheless hair loss 5 years after chemo generic 1 mg finpecia visa, as already mentioned gene therapy hair loss cure discount finpecia 1mg overnight delivery, the knowledgeable clinician should be aware of those diseases that are attributable entirely to a gene mutation as well as the ongoing endeavors to understand the cellular mechanisms being exposed as a result of the study of genetic forms of neurologic disease hair loss in men over 30 cheap finpecia 1 mg on line. In respect to their temporal evolution hair loss itchy scalp buy finpecia australia, these diseases differ from most of the metabolic and slow viral disorders. Often a skillfully taken history will reveal that the patient or family had been aware of a pre-existent condition, the subtle symptoms of which had been present for some time but had attracted little attention. Whether trauma or other stress can actually evoke or aggravate a degenerative disease is a question that cannot be answered with absolute certainty. Anyone who states otherwise must offer evidence that at present is purely anecdotal. Instead, these degenerative disease processes by their very nature appear to develop de novo, without relation to known antecedent events, and their symptomatic expressions are late events in the pathologic process, occurring only when the degree of neuronal loss reaches or exceeds the "safety factor" for the functioning of a particular neuronal system. Irreversibility of clinical manifestations is another feature common to all the neurodegenerative conditions. The familial occurrence of disease is of great importance both clinically and for scientific reasons, as just mentioned, but it must be emphasized again that such information is often difficult to obtain on first contact with the patient. The family may be small or widely scattered, so that the patient is unaware of the health of other members. Furthermore, it may not be realized that an illness is hereditary if other members of the family have a much more or much less severe form of the disorder than the patient. Sometimes, in the latter case, only the careful examination of other family members will disclose the presence of a hereditary disease. Also, it should be remembered that familial occurrence of a disease does not necessarily mean that it is inherited but may indicate instead that more than one member of a family had been exposed to the same infectious or toxic agent. As a rule, the degenerative diseases of the nervous system run a ceaselessly progressive course and with few exceptions are uninfluenced by any medical or surgical measures, so that dealing with a patient with this type of illness may be an anguishing experience for all concerned. However, some of these diseases are characterized by periods of relative stability; moreover, many symptoms. General Pathologic and Pathogenic Features Most of the degenerative diseases are characterized by the selective involvement of anatomically and physiologically related systems of neurons. These degenerative diseases have therefore been called system atrophies or systemic neuronal atrophies. The selective vulnerability of certain systems of neurons is not an exclusive property of the degenerative diseases; several different disease processes of known cause have similarly circumscribed effects on the nervous system. Diphtheria toxin, for instance, selectively affects the myelin of the peripheral nerves near the spinal ganglia, and triorthocresyl phosphate affects both the corticospinal tracts of the spinal cord and the spinal motor neurons. Other examples are the special vulnerability of the Purkinje cells to hyperthermia, the cerebellar granule cells to methyl mercury compounds, the basal ganglionic neurons to manganese, and the hippocampal neurons to anoxia. On the other hand, in Alzheimer disease and some other degenerative diseases, the pathologic changes are somewhat less selective and eventually more diffuse, but still restricted largely to neurons in the cerebral cortex. Even then, there is an early proclivity to involve certain neurons, namely those of the hippocampus and related structures. As one would expect of any pathologic process that is based on the slow wasting and loss of neurons, not only the cell bodies but also their dendrites, axons, and myelin sheaths disappear- unaccompanied by an intense tissue reaction or cellular response because of the slowness of the process. These radiologic findings help distinguish the neuronal atrophies from other large classes of progressive disease of the nervous system- namely, tumors, infections, and other processes of inflammatory type. The terms atrophy and degeneration describe phenomena that are manifest in systems or subsets of neurons, and they apply to the entire class of degenerative diseases of the nervous system, both sporadic and genetic types. At the cellular level, several processes characterize the death of individual cells. The term apoptosis has been borrowed from embryology to specify many of the diverse mechanisms that lead to neuronal degeneration. The original meaning of the term refers to the naturally occurring cell death during development and involves the expression of genes that cause a reduction in the number of neurons over a short period of time. The process of pathologic neuronal degeneration is quite different in that it refers to a series of changes in mature neurons that occur over a protracted period of time, leading to cell death and often leaving a discrete glial scar. In many models, this process can involve activation of programmed cell death genes, although the time course and cellular morphology are not apoptotic in the original sense of the term. Probably, mechanisms other than programmed cell death will prove central to understanding the degenerative diseases, and it is becoming apparent that the clinical features of these conditions are manifest even before cellular destruction occurs. For example, interference with synaptic signaling and dysfunction of supporting glial cells are equally important features.
Fourth hair loss in men 80s clothing buy 1 mg finpecia overnight delivery, if the congenital abnormality has occurred in other members of the family of the same or previous generations hair loss in men whom men buy cheap finpecia 1 mg online, it is usually genetic- although hair loss guinea pigs order finpecia online now, as noted above hair loss male forum proven 1mg finpecia, this does not exclude the possible adverse effects of exogenous agents. Fifth, many of the teratologic conditions that cause birth defects pass unrecognized because they end in spontaneous abortions. Sixth, low birth weight and gestational age, indicative of premature birth, increase the risk of mental subnormality, seizures, cerebral palsy, and death. Regarding etiology, which is really the crux of the problem of birth defects, some order and classification have emerged. In general, malformations may be subdivided into four groups: (1) one in which a single mutant gene is responsible (2. It has been stated that true malformations are due to fundamental endogenous disturbances of cytogenesis and histogenesis occurring in the first half of gestation and that exogenous factors, which destroy brain tissue but do not cause malformations, operate in the second half. An exogenous lesion occurring during the embryonal period may not only destroy tissue but also derail the neuronal migrations of normal development. Cephalic and spinal meningocele, meningoencephalocele, Dandy-Walker syndrome, meningomyelocele 2. Other restricted congenital abnormalities (Horner syndrome, unilateral ptosis, anisocoria, etc. Congenital extrapyramidal disorders (double athetosis; erythroblastosis fetalis and kernicterus) E. Mental retardation A textbook on principles of neurology is not the place in which to present a detailed account of all the hereditary and congenital developmental abnormalities that might affect the nervous system. For such details, the interested reader should refer to several excellent monographs. These are supplemented by special atlases of congenital malformations mentioned further on. In this chapter we sketch only the major groups and discuss in detail a few of the more common disease entities. The classification in Table 38-1 adheres to a grouping in accordance with the main presenting abnormality or abnormalities. Represented here are the common problems that lead families to seek consultation with the pediatric neurologist: (1) structural defects of the cranium, spine, and limbs, and of eyes, nose, ears, jaws, and skin; (2) disturbed motor function- taking the form of retarded development or abnormal movements; (3) epilepsy; and (4) mental retardation. One has only to walk through an institution for the mentally retarded to appreciate the remarkable number and diversity of physical disfigurements that attend abnormalities of the nervous system. Smith, in the third edition of his monograph on the patterns of human malformations, listed 345 distinctive syndromes; in the fourth edition (edited by K. Indeed, a normal-appearing and severely retarded individual stands out in such a crowd and will frequently be found to have an inherited metabolic defect or birth injury. The intimate relationship between the growth and development of the cranium and that of the brain deserves comment. In embryonic life the most rapidly growing parts of the neural tube induce special changes in and at the same time are influenced by the overlying mesoderm (a process known as induction); hence abnormalities in the formation of skull, orbits, nose, and spine are regularly associated with anomalies of the brain and spinal cord. During early fetal life the cranial bones and vertebral arches enclose and protect the developing brain and spinal cord; throughout the period of rapid brain growth, as pressure is exerted on the inner table of the skull, the latter accommodates to the increasing size of the brain. This adaptation is facilitated by the membranous fontanels, which remain open until maximal brain growth has been attained; only then do they ossify (close). In addition, stature is controlled by the nervous system, as shown by the fact that a majority of mentally retarded individuals are also stunted physically to a varying degree. Cranial Malformations at Birth and in Early Infancy Certain alterations in size and shape of the head in the infant, child, or even adult always signify a pathologic process that affected the brain before birth or in early infancy. The size of the cranium reflects the size of the brain; therefore the tape measure is one of the most useful tools in pediatric neurology- no examination in a neurologically affected child is complete without a measurement of the circumference of the head. Graphs of the head circumference in males and females from birth to 18 years of age have been compiled by Nellhaus.
Cheap finpecia line. What is PCOS in WOMEN? How to cure PCOS naturally?(EXPLAINED!!).
Dopamine D2-like receptors are expressed in pancreatic beta cells and mediate inhibition of insulin secretion hair loss in men 3 button purchase finpecia 1mg with visa. Proteins associated with immunopurified granules from a model pancreatic islet beta-cell system: proteomic snapshot of an endocrine secretory granule hair loss blogs purchase finpecia. Microtubules and beta cell function: effect of colchicine on microtubules and insulin secretion in vitro by mouse beta cells hair loss zoladex quality 1 mg finpecia. Suppression of the expression of a pancreatic beta-cell form of the kinesin heavy chain by antisense oligonucleotides inhibits insulin secretion from primary cultures of mouse beta-cells hair loss in men 30 buy finpecia with a mastercard. Molecular mechanisms involved in secretory vesicle recruitment to the plasma membrane in beta-cells. Kinectin participates in microtubule-dependent hormone secretion in pancreatic islet beta-cells. Expression and localisation of synaptotagmin isoforms in endocrine beta-cells: their function in insulin exocytosis. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Fast exocytosis with few Ca2+ channels in insulin-secreting mouse pancreatic B cells. Protein kinases, protein phosphorylation, and the regulation of insulin secretion from pancreatic beta-cells. Dominance of cyclooxygenase-2 in the regulation of pancreatic islet prostaglandin synthesis. Expression and function of cyclooxygenase and lipoxygenase enzymes in human islets of Langerhans. Protein kinase C in beta-cells: expression of multiple isoforms and involvement in cholinergic stimulation of insulin secretion. Glucose-stimulated insulin secretion is not dependent on activation of protein kinase A. Glucose controls cytosolic Ca2+ and insulin secretion in mouse islets lacking adenosine triphosphate-sensitive K+ channels owing to a knockout of the pore-forming subunit Kir6. Two sites of glucose control of insulin release with distinct dependence on the energy state in pancreatic B-cells. Cyclic adenosine monophosphate differently affects the response of mouse pancreatic beta-cells to various amino acids. Identification of insulin signaling elements in human beta-cells: autocrine regulation of insulin gene expression. Effects of glucose, exogenous insulin, and carbachol on C-peptide and insulin secretion from isolated perifused rat islets. Selective insulin signaling through A and B insulin receptors regulates transcription of insulin and glucokinase genes in pancreatic beta cells. Autocrine anti-apoptotic and proliferative effects of insulin in pancreatic beta-cells. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Glucose inhibits glucagon secretion by a direct effect on mouse pancreatic alpha cells. Somatostatin inhibits insulin and glucagon secretion via two receptors subtypes: an in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Suppression of insulin release by galanin and somatostatin is mediated by a G-protein: an effect involving repolarization and reduction in cytoplasmic free Ca2+ concentration. Effects of rat pancreatic polypeptide on islet-cell secretion in the perfused rat pancreas.

Muscles of facial expression hair loss cure natural way discount 1 mg finpecia with amex, mastication hair loss medication results order 1mg finpecia with amex, swallowing hair loss zurich buy finpecia 1 mg line, and speech are affected in 80 percent of patients at some time in the illness hair loss in men 90s fashion discount 1 mg finpecia amex, and in 5 to 10 percent these are the first or only muscles to be involved. Less frequent is an initial or early involvement of the flexors and extensors of the neck, muscles of the shoulder girdle, and flexors of the hips. In the most advanced cases, all muscles are weakened, including the diaphragmatic, abdominal, and intercostal muscles and even the external sphincters of the bladder and bowel. The involvement of any group of muscles closely parallels their degree of weakness early in the disease. The clinical rule holds that the proximal muscles are far more vulnerable than distal ones, as they are in almost all other forms of myopathy. The other characteristic feature of myasthenic weakness is its tendency to increase as the day wears on or with repeated use of an affected muscle group, but patients seldom volunteer this information. A few patients report being paradoxically worse on awakening, especially if they have not received medication during the night. In broad terms, therefore, myasthenia gravis may be conceived as a fluctuating and fatigable oculofaciobulbar palsy. To reiterate the topographic attributes of the illness, combined weakness of the extraocular muscles, levators, and orbicularis oculi combined with normal pupillary responses to light and accommodation is virtually diagnostic of myasthenia, especially if the function of these muscles is restored after a period of rest. It may be more difficult to eat after talking, and the voice fades and becomes nasal after sustained conversation. Women may complain of inability to fix their hair or makeup because of fatigue of the shoulders, or of difficulty in applying lipstick because they are unable to purse and roll their lips. In cases with generalized weakness, there may be difficulty in retaining flatus because of weakness of the external sphincter. A temporary increase in weakness may follow vaccination, menstruation, and exposure to extremes of temperature. A peculiarity of myasthenic muscle contraction that may be observed occasionally is a sudden lapse of sustained posture or interruption of movement resulting in a kind of irregular tremor, similar to that of normal muscle nearing the point of exhaustion. A dynamometer demonstrates the rapidly waning power of contraction of a series of hand grips, and repetitive stimulation of a motor nerve at slow rates while recording muscle action potentials the same decremental strength in a more quantitative fashion (see. Weakened muscles in myasthenia gravis undergo atrophy to only a minimal degree or not at all. Even repeated tapping of a tendon does not usually tax muscles to the point where contraction fails. Smooth and cardiac muscles are not involved and other neural functions are preserved. Weakened muscles, especially those of the eyes and back of the neck, may ache, but pain is seldom an important complaint. Paresthesias of the face, hands, and thighs are reported infrequently but are not accompanied by demonstrable sensory loss. Anosmia and ageusia have been mentioned as rare findings, but they may be coincidental. The tongue may display one central and two lateral longitudinal furrows (trident tongue), as pointed out originally by Buzzard. Its prevalence is variously estimated to be from 43 to 84 per million of the population and the annual incidence rate, approximately 1 per 300,000. The disease may begin at any age, but onset in the first decade is relatively rare (only 10 percent of cases occur under the age of 10 years). The peak age of onset is between 20 and 30 years in women and between 50 and 60 years in men. Under the age of 40, females are affected two to three times as often as males, whereas in later life, the incidence in males is higher (3:2). Of patients with thymomas, the majority are older (50 to 60 years), and males predominate. To facilitate clinical staging of therapy and prognosis, the following classification, introduced by Osserman, remains useful (the relative incidence of each type is indicated and "drug response" refers to treatment with an anticholinesterase): I.